

Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Cytoskeleton and Its Associated Proteins Involved in Mechanotransduction
3. Alterations in Cells’ Physical Properties Promote the Creation of a Procancer Microenvironment
4. New Cancer Genes Defined by Dysregulations in Mechanotransduction
5. Epithelial–Mesenchymal Transition (EMT) Induction by Deformations of Cytoskeleton and the Associated Proteins
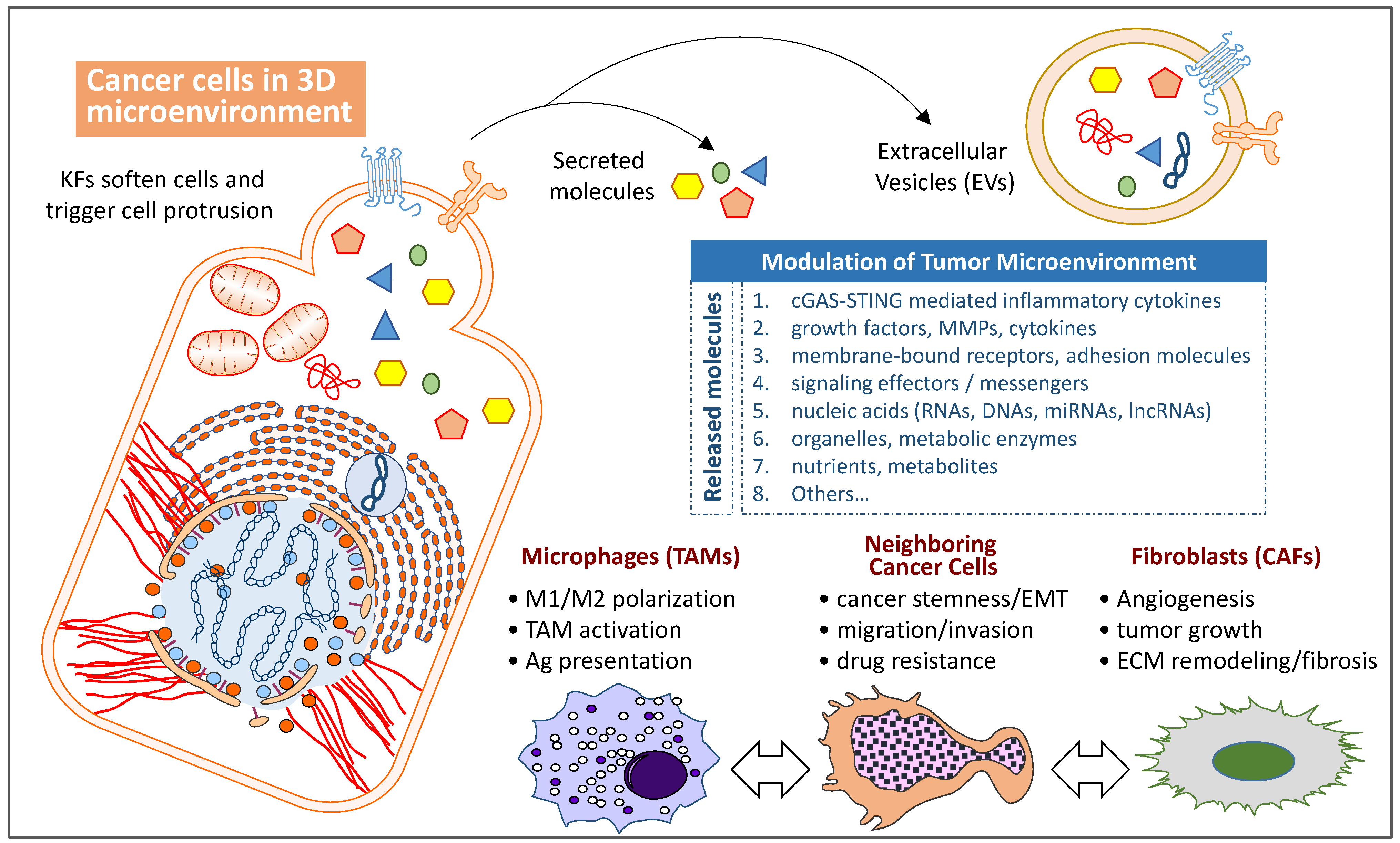
6. Regulation of Immune TME by Mutations in Cytoskeleton-Associated Proteins
7. ECM Stiffening Reprograms Cancer Cell Metabolism Leading to Autophagy-Mediated Clonal Selection/Evolution
8. Seesaw Effects between PFKFB3 and Autophagy
9. Roles of pEMT and the Tumor Microenvironment in Tumor Progression
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Majeski, H.E.; Yang, J. The 2016 John J. Abel Award Lecture: Targeting the Mechanical Microenvironment in Cancer. Mol. Pharmacol. 2016, 90, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przybyla, L.; Muncie, J.M.; Weaver, V.M. Mechanical Control of Epithelial-to-Mesenchymal Transitions in Development and Cancer. Annu. Rev. Cell Dev. Biol. 2016, 32, 527–554. [Google Scholar] [CrossRef] [PubMed]
- Ciasca, G.; Papi, M.; Minelli, E.; Palmieri, V.; De Spirito, M. Changes in cellular mechanical properties during onset or progression of colorectal cancer. World J. Gastroenterol. 2016, 22, 7203–7214. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, S. Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1209–H1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Chicurel, M.E.; Chicurel, M.E.; Singer, R.H.; Meyer, C.J.; Ingber, D.E. Integrin binding and mechanical tension induce movement of mRNA and ribosomes to focal adhesions. Nature 1998, 392, 730–733. [Google Scholar] [CrossRef]
- Vining, K.H.; Mooney, D.J. Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol. 2017, 18, 728–742. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.L.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef]
- Nia, H.T.; Munn, L.L.; Jain, R.K. Physical traits of cancer. Science 2020, 370, eaaz0868. [Google Scholar] [CrossRef]
- Benham-Pyle, B.W.; Pruitt, B.L.; Nelson, W.J. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and β-catenin activation to drive cell cycle entry. Science 2015, 348, 1024–1027. [Google Scholar] [CrossRef] [Green Version]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.L.; Gourdon, D.; Little, W.C.; Kubow, K.E.; Eguiluz, R.A.; Luna-Morris, S.; Vogel, V. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 2007, 5, e268. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.; Cho, S.; Dooling, L.J.; Discher, D.E. Tension in fibrils suppresses their enzymatic degradation—A molecular mechanism for ‘use it or lose it’. Matrix Biol. 2020, 85–86, 34–46. [Google Scholar] [CrossRef]
- Kubow, K.E.; Vukmirovic, R.; Zhe, L.; Klotzsch, E.; Smith, M.L.; Gourdon, D.; Luna, S.; Vogel, V. Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix. Nat. Commun. 2015, 6, 8026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer. 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer. 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.; Guan, K.L. The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 216, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [Green Version]
- Helmlinger, G.; Netti, P.A.; Lichtenbeld, H.C.; Melder, R.J.; Jain, R.K. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 1997, 15, 778–783. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, L.L.; Jain, R.K. Vascular regulation of antitumor immunity. Science 2019, 365, 544–545. [Google Scholar] [CrossRef] [PubMed]
- Taheri, F.; Isbilir, B.; Müller, G.; Krieger, J.W.; Chirico, G.; Langowski, J.; Tóth, K. Random Motion of Chromatin Is Influenced by Lamin A Interconnections. Biophys. J. 2018, 114, 2465–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskratsch, T.; Wolfenson, H.; Sheetz, M.P. Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 2014, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef]
- Tse, J.M.; Cheng, G.; Tyrrell, J.A.; Wilcox-Adelman, S.A.; Boucher, Y.; Jain, R.K.; Munn, L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 911–916. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Sánchez, M.E.; Barbier, S.; Whitehead, J.; Béalle, G.; Michel, A.; Latorre-Ossa, H.; Rey, C.; Fouassier, L.; Claperon, A.; Brullé, L.; et al. Mechanical induction of the tumorigenic β-catenin pathway by tumour growth pressure. Nature 2015, 523, 92–95. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Luo, Q.; Ju, Y.; Song, G. A Soft Matrix Enhances the Cancer Stem Cell Phenotype of HCC Cells. Int. J. Mol. Sci. 2019, 20, 2831. [Google Scholar] [CrossRef]
- Swaminathan, V.; Mythreye, K.; O’Brien, E.T.; Berchuck, A.; Blobe, G.C.; Superfine, R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 2011, 71, 5075–5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, S.E.; Jin, Y.S.; Rao, J.; Gimzewski, J.K. Nanomechanical analysis of cells from cancer patients. Nat. Nanotechnol. 2007, 2, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Tajik, A.; Chen, J.; Jia, Q.; Chowdhury, F.; Wang, L.; Chen, J.; Zhang, S.; Hong, Y.; Yi, H.; et al. Matrix softness regulates plasticity of tumour-repopulating cells via H3K9 demethylation and Sox2 expression. Nat. Commun. 2014, 5, 4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.H.; Lin, H.K.; Lin, I.H.; Chiou, Y.W.; Chen, H.W.; Liu, C.Y.; Harn, H.I.; Chiu, W.T.; Wang, Y.K.; Shen, M.R.; et al. Mechanical phenotype of cancer cells: Cell softening and loss of stiffness sensing. Oncotarget 2015, 6, 20946–20958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.L.; Pegoraro, A.F.; Li, H.; Li, K.; Yuan, Y.; Xu, G.; Gu, Z.; Sun, J.; Hao, Y.; Gupta, S.K.; et al. Cell swelling, softening and invasion in a three-dimensional breast cancer model. Nat. Phys. 2020, 16, 101–108. [Google Scholar] [CrossRef]
- Kumar, R.; Sanawar, R.; Li, X.; Li, F. Structure, biochemistry, and biology of PAK kinases. Gene 2017, 605, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Hourwitz, M.J.; Campanello, L.; Fourkas, J.T.; Losert, W.; Parent, C.A. Actin Cytoskeleton and Focal Adhesions Regulate the Biased Migration of Breast Cancer Cells on Nanoscale Asymmetric Sawteeth. ACS Nano 2019, 13, 1454–1468. [Google Scholar] [CrossRef]
- Gkretsi, V.; Stylianopoulos, T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front. Oncol. 2018, 8, 145. [Google Scholar] [CrossRef]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate Filaments at the Junction of Mechanotransduction, Migration, and Development. Front. Cell Dev. Biol. 2017, 5, 81. [Google Scholar] [CrossRef] [Green Version]
- Laly, A.C.; Sliogeryte, K.; Pundel, O.J.; Ross, R.; Keeling, M.C.; Avisetti, D.; Waseem, A.; Gavara, N.; Connelly, J.T. The keratin network of intermediate filaments regulates keratinocyte rigidity sensing and nuclear mechanotransduction. Sci. Adv. 2021, 7, eabd6187. [Google Scholar] [CrossRef]
- Tsai, F.J.; Lai, M.T.; Cheng, J.; Chao, S.C.; Korla, P.K.; Chen, H.J.; Lin, C.M.; Tsai, M.H.; Hua, C.H.; Jan, C.I.; et al. Novel K6-K14 75 enhances cancer stemness and aggressiveness in oral squamous cell carcinoma. Oncogene 2019, 38, 5113–5126. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Paul, M.K.; Alioscha-Perez, M.; Grosberg, A.; Sahli, H.; Dubinett, S.M.; Weiss, S. Statistical parametrization of cell cytoskeleton reveals lung cancer cytoskeletal phenotype with partial EMT signature. Commun. Biol. 2022, 5, 407. [Google Scholar] [CrossRef] [PubMed]
- Ampe, C.; Witjes, L.; Van Troys, M. Cancer type-specific alterations in actin genes: Worth a closer look? Int. Rev. Cell Mol. Biol. 2021, 360, 133–184. [Google Scholar] [PubMed]
- Suresh, R.; Diaz, R.J. The remodelling of actin composition as a hallmark of cancer. Transl. Oncol. 2021, 14, 101051. [Google Scholar] [CrossRef]
- Drukman, S.; Kavallaris, M. Microtubule alterations and resistance to tubulin-binding agents (review). Int. J. Oncol. 2002, 21, 621–628. [Google Scholar] [CrossRef]
- Verrills, N.M.; Flemming, C.L.; Liu, M.; Ivery, M.T.; Cobon, G.S.; Norris, M.D.; Haber, M.; Kavallaris, M. Microtubule alterations and mutations induced by desoxyepothilone B: Implications for drug-target interactions. Chem. Biol. 2003, 10, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Nami, B.; Wang, Z. Genetics and Expression Profile of the Tubulin Gene Superfamily in Breast Cancer Subtypes and Its Relation to Taxane Resistance. Cancers 2018, 10, 274. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef] [Green Version]
- Hassan Ibrahim, I.; Balah, A.; Gomaa Abd Elfattah Hassan, A.; Gamal Abd El-Aziz, H. Role of motor proteins in human cancers. Saudi J. Biol. Sci. 2022, 29, 103436. [Google Scholar] [CrossRef]
- Wattanathamsan, O.; Pongrakhananon, V. Emerging role of microtubule-associated proteins on cancer metastasis. Front. Pharmacol. 2022, 13, 935493. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Raphael, B.J.; Dobson, J.R.; Oesper, L.; Vandin, F. Identifying driver mutations in sequenced cancer genomes: Computational approaches to enable precision medicine. Genome Med. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzetta, V.; Musella, I.; Rapa, I.; Volante, M.; Netti, P.A.; Fusco, S. Mechanical phenotyping of cells and extracellular matrix as grade and stage markers of lung tumor tissues. Acta Biomater. 2017, 57, 334–341. [Google Scholar] [CrossRef]
- Angel, P.M.; Bruner, E.; Bethard, J.; Clift, C.L.; Ball, L.; Drake, R.R.; Feghali-Bostwick, C. Extracellular matrix alterations in low-grade lung adenocarcinoma compared with normal lung tissue by imaging mass spectrometry. J. Mass Spectrom. 2020, 55, e4450. [Google Scholar] [CrossRef]
- Cochlin, D.L.; Ganatra, R.H.; Griffiths, D.F. Elastography in the detection of prostatic cancer. Clin. Radiol. 2002, 57, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.F.; Li, Q.; Melnichouk, O.; Huszti, E.; Martin, L.J.; Gunasekara, A.; Mawdsley, G.; Yaffe, M.J.; Minkin, S. Evidence that breast tissue stiffness is associated with risk of breast cancer. PLoS ONE 2014, 9, e100937. [Google Scholar] [CrossRef] [PubMed]
- Maskarinec, G.; Pagano, I.S.; Little, M.A.; Conroy, S.M.; Park, S.Y.; Kolonel, L.N. Mammographic density as a predictor of breast cancer survival: The Multiethnic Cohort. Breast Cancer Res. 2013, 15, R7. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knights, A.J.; Funnell, A.P.; Crossley, M.; Pearson, R.C. Holding Tight: Cell Junctions and Cancer Spread. Trends Cancer Res. 2012, 8, 61–69. [Google Scholar]
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Investig. 2009, 119, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Leggett, S.E.; Hruska, A.M.; Guo, M.; Wong, I.Y. The epithelial-mesenchymal transition and the cytoskeleton in bioengineered systems. Cell Commun. Signal. 2021, 19, 32. [Google Scholar] [CrossRef]
- Le Maout, E.; Lo Vecchio, S.; Kumar Korla, P.; Jinn-Chyuan Sheu, J.; Riveline, D. Ratchetaxis in Channels: Entry Point and Local Asymmetry Set Cell Directions in Confinement. Biophys. J. 2020, 119, 1301–1308. [Google Scholar] [CrossRef]
- Reinholz, M.M.; Kitzmann, K.A.; Tenner, K.; Hillman, D.; Dueck, A.C.; Hobday, T.J.; Northfelt, D.W.; Moreno-Aspitia, A.; Roy, V.; LaPlant, B.; et al. Cytokeratin-19 and mammaglobin gene expression in circulating tumor cells from metastatic breast cancer patients enrolled in North Central Cancer Treatment Group trials, N0234/336/436/437. Clin. Cancer Res. 2011, 17, 7183–7193. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Yasuchika, K.; Ishii, T.; Katayama, H.; Yoshitoshi, E.Y.; Ogiso, S.; Kita, S.; Yasuda, K.; Fukumitsu, K.; Mizumoto, M.; et al. Keratin 19, a cancer stem cell marker in human hepatocellular carcinoma. Clin. Cancer Res. 2015, 21, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.; Wang, J.; Mancianti, M.L.; Epstein, E.H., Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1+/− mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, W.; Chen, J.; Morrison, G.D.; Huang, S.; Creighton, C.J.; Huang, J.; Chamness, G.C.; Hilsenbeck, S.G.; Roop, D.R.; Leavitt, A.D.; et al. Keratin 6a marks mammary bipotential progenitor cells that can give rise to a unique tumor model resembling human normallike breast cancer. Oncogene 2011, 30, 4399–4409. [Google Scholar] [CrossRef] [Green Version]
- Ujiie, D.; Okayama, H.; Saito, K.; Ashizawa, M.; Thar Min, A.K.; Endo, E.; Kase, K.; Yamada, L.; Kikuchi, T.; Hanayama, H.; et al. KRT17 as a prognostic biomarker for stage II colorectal cancer. Carcinogenesis 2020, 41, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, S.; Kim, N.W.; Wu, M.; Chan, I.; Escobar-Hoyos, L.F.; Shroyer, K.R. Keratin 17 Is a Novel Cytologic Biomarker for Urothelial Carcinoma Diagnosis. Am. J. Clin. Pathol. 2021, 156, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, Y.; Velculescu, V.E. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004, 3, 1221–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, A.B.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998, 58, 1130–1134. [Google Scholar]
- de La Coste, A.; Romagnolo, B.; Billuart, P.; Renard, C.A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef] [Green Version]
- Gracilla, D.E.; Korla, P.K.; Lai, M.T.; Chiang, A.J.; Liou, W.S.; Sheu, J.J. Overexpression of wild type or a Q311E mutant MB21D2 promotes a pro-oncogenic phenotype in HNSCC. Mol. Oncol. 2020, 14, 3065–3082. [Google Scholar] [CrossRef]
- Chow, K.L.; Hall, D.H.; Emmons, S.W. The mab-21 gene of Caenorhabditis elegans encodes a novel protein required for choice of alternate cell fates. Development 1995, 121, 3615–3626. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Mizutani-Koseki, Y.; Koseki, H.; Takahashi, N. Requirement for Mab21l2 during development of murine retina and ventral body wall. Dev. Biol. 2004, 274, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, K.; Knizewski, L.; Wyrwicz, L.S.; Rychlewski, L.; Ginalski, K. Comprehensive classification of nucleotidyltransferase fold proteins: Identification of novel families and their representatives in human. Nucleic Acids Res. 2009, 37, 7701–7714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira Mann, C.C.; Kiefersauer, R.; Witte, G.; Hopfner, K.-P. Structural and biochemical characterization of the cell fate determining nucleotidyltransferase fold protein MAB21L1. Sci. Rep. 2016, 6, 27498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rossum, D.B.; Patterson, R.L.; Cheung, K.-H.; Barrow, R.K.; Syrovatkina, V.; Gessell, G.S.; Burkholder, S.G.; Watkins, D.N.; Foskett, J.K.; Snyder, S.H. DANGER, a novel regulatory protein of inositol 1,4,5-trisphosphate-receptor activity. J. Biol. Chem. 2006, 281, 37111–37116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Wu, X.; Wu, F.-H.; Wang, X.; Wang, L.; Siedow, J.N.; Zhang, W.; Pei, Z.-M. Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STING. Nucleic Acids Res. 2014, 42, 8243–8257. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Mak, M.; Spill, F.; Kamm, R.D.; Zaman, M.H. Single-Cell Migration in Complex Microenvironments: Mechanics and Signaling Dynamics. J. Biomech. Eng. 2016, 138, 021004. [Google Scholar] [CrossRef] [Green Version]
- Spatarelu, C.P.; Zhang, H.; Trung Nguyen, D.; Han, X.; Liu, R.; Guo, Q.; Notbohm, J.; Fan, J.; Liu, L.; Chen, Z. Biomechanics of Collective Cell Migration in Cancer Progression: Experimental and Computational Methods. ACS Biomater. Sci. Eng. 2019, 5, 3766–3787. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.J.; Tang, Y.J.; Tang, Y.L.; Liang, X.H. What makes cells move: Requirements and obstacles for leader cells in collective invasion. Exp. Cell Res. 2019, 382, 111481. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Saha, P.; Samanta, A.; Bishayee, A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules 2020, 10, 1561. [Google Scholar] [CrossRef]
- De Pascalis, C.; Etienne-Manneville, S. Single and collective cell migration: The mechanics of adhesions. Mol. Biol. Cell. 2017, 28, 1833–1846. [Google Scholar] [CrossRef]
- van Helvert, S.; Storm, C.; Friedl, P. Mechanoreciprocity in cell migration. Nat. Cell Biol. 2018, 20, 8–20. [Google Scholar] [CrossRef]
- Wilhelmsen, K.; Litjens, S.H.; Sonnenberg, A. Multiple functions of the integrin alpha6beta4 in epidermal homeostasis and tumorigenesis. Mol. Cell Biol. 2006, 26, 2877–2886. [Google Scholar] [CrossRef] [Green Version]
- Danen, E.H.; Sonneveld, P.; Brakebusch, C.; Fassler, R.; Sonnenberg, A. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J. Cell Biol. 2002, 159, 1071–1086. [Google Scholar] [CrossRef] [Green Version]
- Heino, J. The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biol. 2000, 19, 319–323. [Google Scholar] [CrossRef]
- Lai, M.T.; Hua, C.H.; Tsai, M.H.; Wan, L.; Lin, Y.J.; Chen, C.M.; Chiu, I.W.; Chan, C.; Tsai, F.J.; Sheu, J.J.C. Talin-1 overexpression defines high risk for aggressive oral squamous cell carcinoma and promotes cancer metastasis. J. Pathol. 2011, 224, 367–376. [Google Scholar] [CrossRef]
- Jin, J.K.; Tien, P.C.; Cheng, C.J.; Song, J.H.; Huang, C.; Lin, S.H.; Gallick, G.E. Talin1 phosphorylation activates β1 integrins: A novel mechanism to promote prostate cancer bone metastasis. Oncogene 2015, 34, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Deb, B.; Puttamallesh, V.N.; Gondkar, K.; Thiery, J.P.; Gowda, H.; Kumar, P. Phosphoproteomic Profiling Identifies Aberrant Activation of Integrin Signaling in Aggressive Non-Type Bladder Carcinoma. J. Clin. Med. 2019, 8, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Lykov, N.; Tzeng, C. Talin-1 interaction network in cellular mechanotransduction (Review). Int. J. Mol. Med. 2022, 49, 60. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ghosh, S.; Wang, Z.; Hunter, T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4, 499–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, S.; Doi, R.; Toyoda, E.; Tsuji, S.; Wada, M.; Koizumi, M.; Tulachan, S.S.; Ito, D.; Kami, K.; Mori, T.; et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 2004, 10, 4125–4133. [Google Scholar] [CrossRef] [Green Version]
- Korla, P.K.; Chen, C.C.; Gracilla, D.E.; Lai, M.T.; Chen, C.M.; Chen, H.Y.; Hwang, T.; Chen, S.Y.; Sheu, J.J. Somatic mutational landscapes of adherens junctions and their functional consequences in cutaneous melanoma development. Theranostics 2020, 10, 12026–12043. [Google Scholar] [CrossRef]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, Ş.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Yan, X.; Song, C.; Wang, Q.; Wang, Y.; Liu, X.P.; Huang, J.; Li, S.; Hu, W. FAT3 Mutation Is Associated With Tumor Mutation Burden and Poor Prognosis in Esophageal Cancer. Front. Oncol. 2021, 11, 603660. [Google Scholar] [CrossRef]
- Feng, Z.; Yin, Y.; Liu, B.; Zheng, Y.; Shi, D.; Zhang, H.; Qin, J. Prognostic and Immunological Role of FAT Family Genes in Non-Small Cell Lung Cancer. Cancer Control 2022, 29, 10732748221076682. [Google Scholar] [CrossRef]
- Hayashi, M.T.; Karlseder, J. DNA damage associated with mitosis and cytokinesis failure. Oncogene 2013, 32, 4593–4601. [Google Scholar] [CrossRef] [Green Version]
- Garribba, L.; Santaguida, S. The Dynamic Instability of the Aneuploid Genome. Front. Cell Dev. Biol. 2022, 10, 838928. [Google Scholar] [CrossRef]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasugi, M.; Yoshida, Y.; Ohtani, N. Cellular senescence and the tumour microenvironment. Mol Oncol. 2022, 16, 3333–3351. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Peng, P.; Tang, Z.; Zhao, J.; Wu, W.; Li, H.; Shao, M.; Li, L.; Yang, C.; Duan, F.; et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci. Rep. 2017, 7, 39858. [Google Scholar] [CrossRef] [Green Version]
- Weber, G.F.; Bjerke, M.A.; DeSimone, D.W. A mechanoresponsive cadherin-keratin complex directs polarized protrusive behavior and collective cell migration. Dev. Cell 2012, 22, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Zhang, H.; Landmann, F.; Zahreddine, H.; Rodriguez, D.; Koch, M.; Labouesse, M. A tension-induced mechanotransduction pathway promotes epithelial morphogenesis. Nature 2011, 471, 99–103. [Google Scholar] [CrossRef]
- Osmani, N.; Labouesse, M. Remodeling of keratin-coupled cell adhesion complexes. Curr. Opin. Cell Biol. 2015, 32, 30–38. [Google Scholar] [CrossRef]
- Runel, G.; Lopez-Ramirez, N.; Chlasta, J.; Masse, I. Biomechanical Properties of Cancer Cells. Cells 2021, 10, 887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H.; Yingbo, M.; Hangyu, L. The role of YAP/TAZ activity in cancer metabolic reprogramming. Mol. Cancer 2018, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Tian, M.; Pei, Q.; Tan, F.; Pei, H. Extracellular Matrix Stiffness: New Areas Affecting Cell Metabolism. Front. Oncol. 2021, 11, 631991. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Lee, S.B.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.M.; Dayem, A.A.; Cho, S.G. Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. Int. J. Mol. Sci. 2017, 18, 1544. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Bakhshandeh, S.; Werner, C.; Fratzl, P.; Cipitria, A. Microenvironment-mediated cancer dormancy: Insights from metastability theory. Proc. Natl. Acad. Sci. USA 2022, 119, e2111046118. [Google Scholar] [CrossRef]
- Ehrig, S.; Schamberger, B.; Bidan, C.M.; West, A.; Jacobi, C.; Lam, K.; Kollmannsberger, P.; Petersen, A.; Tomancak, P.; Kommareddy, K.; et al. Surface tension determines tissue shape and growth kinetics. Sci. Adv. 2019, 5, eaav9394. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [Green Version]
- Walens, A.; Lin, J.; Damrauer, J.S.; McKinney, B.; Lupo, R.; Newcomb, R.; Fox, D.B.; Mabe, N.W.; Gresham, J.; Sheng, Z.; et al. Adaptation and selection shape clonal evolution of tumors during residual disease and recurrence. Nat. Commun. 2020, 11, 5017. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Nam, J.S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Chen, Y.; Zhu, Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget 2017, 8, 62793–62802. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, Y.; Wang, D.; Huang, C.; Marino, D.; Bollt, O.; Wu, C.; Taylor, M.D.; Li, W.; DeNicola, G.M.; et al. Fascin promotes lung cancer growth and metastasis by enhancing glycolysis and PFKFB3 expression. Cancer Lett. 2021, 518, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Kloog, Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res. 2005, 65, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal. Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Zhang, Y.; Sampathkumar, A. Cytoskeleton Architecture Regulates Glycolysis Coupling Cellular Metabolism to Mechanical Cues. Trends Biochem. Sci. 2020, 45, 637–638. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Dandekar, R.C.; Kingaonkar, A.V.; Dhabekar, G.S. Role of macrophages in malignancy. Ann. Maxillofac. Surg. 2011, 1, 150–154. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, E.J.-Y.; Chen, I.-H.; Kuo, B.Y.-T.; Yu, C.-C.; Lai, M.-T.; Lin, J.-T.; Lin, L.Y.-T.; Chen, C.-M.; Hwang, T.; Sheu, J.J.-C. Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development. Biomolecules 2022, 12, 1862. https://doi.org/10.3390/biom12121862
Wang EJ-Y, Chen I-H, Kuo BY-T, Yu C-C, Lai M-T, Lin J-T, Lin LY-T, Chen C-M, Hwang T, Sheu JJ-C. Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development. Biomolecules. 2022; 12(12):1862. https://doi.org/10.3390/biom12121862
Chicago/Turabian StyleWang, Evan Ja-Yang, I-Hsuan Chen, Brian Yu-Ting Kuo, Chia-Cheng Yu, Ming-Tsung Lai, Jen-Tai Lin, Leo Yen-Ting Lin, Chih-Mei Chen, Tritium Hwang, and Jim Jinn-Chyuan Sheu. 2022. "Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development" Biomolecules 12, no. 12: 1862. https://doi.org/10.3390/biom12121862